
When scientists from Human genome project announced the full range of the human genome in 2003 they moved forward. Indeed, if the researchers did indeed have access to the DNA sequence of most of the genes that code for proteins, then about 8% of this genome had not yet been sequenced. Recently, the international Telomere-to-Telomere (T2T) consortium presented for the first time a complete sequence of the human genome, covering each chromosome from end to end without gaps.
The Human Genome Project, or human genome project, is a program launched in late 1988 to establish complete DNA sequencing of the human genome. In 2003, this project was declared complete, making it possible to map 85% of the genes, the remaining 15% being unattainable with the technologies of the time. Most of the unmapped regions at the time were concentrated around telomeres and centromeres. The first are the caps on the ends of the chromosomes, while the latter are the tightly packed midsections of the chromosomes. In 2013, researchers then narrowed that gap to just 8%, but they still couldn’t integrate and place about 200 million base pairs.
It took nearly 100 scientists from the Telomere-to-Telomere (T2T) Consortium to map the entire human genome. The 8% represents the addition of 400 million new letters added to the existing DNA sequence, which is the equivalent of an entire chromosome! This new work has been published in the magazine Science†
A new reference genome for pangenomics
In 2019, Karen Miga, assistant professor of biomolecular engineering at UC Santa Cruz (UCSC), and Adam Phillippy of the National Human Genome Research Institute (NHGRI), organized an international team of scientists – the Telomere-to-Telomere (T2T) consortium – to fill in the missing pieces. The new reference genome, called T2T-CHM13, adds 99 genes that may encode proteins and nearly 2000 candidate genes that require further research. It also fixes thousands of structural errors in the current reference series.
In addition, the T2T-CHM13 sequence has been fully annotated in the UCSC Genome Browser, providing scientists with an efficient way to access and visualize a wealth of information related to genes and other genome elements. Karen Miga explains in a communicated † We wanted to deliver the information in a way that was accessible and familiar to researchers so they could start using it and using all the tools and resources provided by the browser. †
As such, the new T2T reference genome will complement the standard human reference genome known as the Genome Reference Consortium build 38 (GRCh38), which originates from the publicly funded and continuously updated from the first version. This new reference to the human genome is just a starting point. Indeed, David Haussler, director of the UC Santa Cruz Genomics Institute, explains: The next stage is to think that the reference of humanity’s genome is not a unique sequence of the genome. It is a profound transition, heralding a new era in which we will eventually capture human diversity in an unbiased way †
 100vw, 753px”/>
</picture>
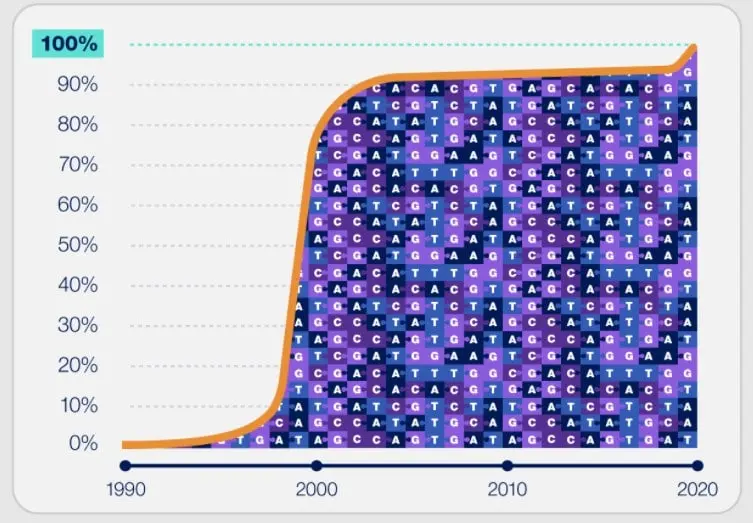
</noscript><figcaption id=) Infographic illustrating that it took almost twice as long to complete the remaining 8% of the human genome than it did to sequence the first 92%. © NHGRIA
Infographic illustrating that it took almost twice as long to complete the remaining 8% of the human genome than it did to sequence the first 92%. © NHGRIAIn this context, the T2T Consortium has joined forces with the Human Pangenome Reference Consortium, which aims to create a new “human pangenome reference” based on the complete genome sequences of 350 individuals. Benedict Paten, associate professor of biomolecular engineering in UCSC’s Baskin School of Engineering, co-author of the paper, says: “Pangenomics is about capturing the diversity of the human population, and it’s also about making sure we’ve captured the entire genome correctly. “. He adds: ” Without having a map of these hard-to-sequence genomic regions across multiple individuals, we’re missing out on much of the variation present in our population. T2T allows us to examine hundreds of genomes, from one telomere to another. It will be great! †
Combining innovative methods for an “incredible scientific success”
To access the missing areas, the team had to use different and finer DNA sequencing methods, which have been implemented in recent years thanks to more modest sequencing costs. Indeed, the cost of sequencing a human genome using so-called “short-read” technologies, which yield hundreds of bases of DNA sequences at a time, is only a few hundred dollars, and has fallen significantly since the end of the human genome project. . However, using these short-readable methods still leaves some gaps in the assembled genomic sequences. With the massive drop in the cost of DNA sequencing comes more investment in new technologies to generate longer reads of DNA sequences without compromising accuracy.
To understand the importance of such reading frames, the authors liken the principle of genome sequencing to cutting a book into pieces of text and then trying to reconstruct it. Sections of text that contain many common or repeated words and phrases are harder to place than unique chunks of text. For example, in the past decade, two new DNA sequencing technologies have emerged, yielding much longer sequence reads: (1) Oxford Nanopore’s “ultra-long” method for sequencing up to 1 million DNA letters with a 5% error rate, in a single read; (2) the PacBio Hifi method of reading letters in packets of 20,000, with near-perfect accuracy – the error rate drops to 0.01%.
Since then, scientists have attempted to further simplify the genome analyzed by these new techniques. As a result, they used an unusual cell type that contains only DNA inherited from the father (most cells in the body contain two genomes — one from each parent), obtained when a sperm fertilizes an egg without a nucleus. In this configuration, the egg is not viable and attaches to the uterus to develop with all the chromosomes from the father but none from the mother. Together, these two advancements have enabled them to decode the more than 3 billion letters that make up the human genome.
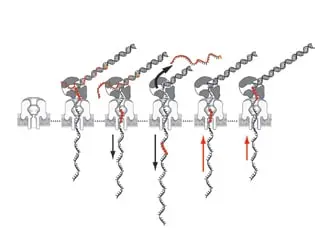
Interestingly, as of 2012, the Oxford Nanopore method relies on nanopore sequencing developed at UC Santa Cruz. The principle is based on the passage of a DNA strand through a nanopore (with a width of only 1.5 nanometers at its narrowest point), formed by a self-assembled protein complex called an ion channel, inserted into a membrane similar to a cell membrane. The membrane separates two wells and an electric field drives the DNA strands (which are negatively charged) from one well to the other through the nanopore. The short blockage of the nanopore by the DNA strands as they pass through the pore produces electrical current modulations that can be analyzed to obtain DNA sequence information.
 100vw, 350px”/>
</picture>
</noscript><figcaption id=) Working principle of nanopores. © Cherf et al., 2012
Working principle of nanopores. © Cherf et al., 2012Oxford Nanopore had the ambition to commercialize this type of sequencing, making it accessible beyond the circle of genomics researchers. Currently, the analysis platform is increasingly used in “applied” contexts such as clinical diagnostics, epidemiology and food safety.
The importance of whole genome knowledge for personalized medicine
According to Evan Eichler, a professor of genome sciences at the University of Washington in Seattle and co-author of the current study, putting the whole puzzle of the human genome together was essential because until now we didn’t know what information came with the missing 8 %. According to the scientists, this complete mapping of the genome will allow a better understanding of our evolution, while enabling medical discoveries in areas such as aging, neurodegenerative diseases, cancer and heart disease.
In addition, some of the extra genes would be those of the immune response, allowing people to adapt to infections and viruses. The other parts of the genome, with many repeats, include those where the most genetic variation is found. Variability within these regions may therefore provide crucial clues as to how our ancestors underwent rapid evolutionary changes, leading to more complex cognition.
In addition, according to Adam Phillippy, co-chair of the consortium, sequencing a person’s entire genome is expected to become cheaper and easier in the coming years. He explains: ” In the future, if someone has their genome sequenced, we can identify all of their DNA variants and use that information to better manage their healthcare. †
Finally, the work is also likely to lead to a better understanding of centromeres. They are dense bundles of DNA that hold chromosomes together and play a role in cell division. Until now, they have been considered unassignable because they contain thousands of DNA sequences that repeat over and over.

